III. Изомерия
Как отмечалось ранее, замечательным достижением теории химического строения А. М. Бутлерова явилось предсказание явления изомерии, которое чрезвычайно характерно для органических соединений и оказывает существенное влияние на их физические и химические свойства. Изомеры – вещества, имеющие одну и ту же брутто-формулу, но отличающиеся между собой строением и свойствами. Известны два основных класса – структурные и пространственные изомеры, которые, в свою очередь, могут подразделяться на различные типы (табл. 3-1).
Таблица 3-1
Типы изомерии
|
Отношение |
Изомерия структурная |
Изомерия |
|
Статическая |
Скелета, |
Геометрическая |
|
Функциональной |
Оптическая |
|
|
Положения |
|
|
|
Динамическая |
Таутомерия |
Конформационная |
*
Функциональная группа – структурный фрагмент
в молекуле, определяющий химические свойства данного класса соединений.
Изомеры по своему поведению в стандартных условиях относятся к статическому или к динамическому ряду. Первые в “нормальных” условиях устойчивы, могут быть выделены и существуют в индивидуальном виде, что обусловлено значительным энергетическим барьером между ними. Взаимопереход связан с разрывом ковалентной связи, прочность которой значительно превосходит возможность “теплового” движения атомов в молекуле.
Вторые находятся в “нормальных” условиях в динамическом равновесии, скорость взаимоперехода изомеров такова, что их невозможно выделить в индивидуальном виде.
Энергетические барьеры между динамическими изомерами столь малы, что легко преодолеваются тепловым движением атомов в молекуле.
3.1. Структурная изомерия
Соединения в парах а, б, в, г, д являются структурными изомерами, то есть отличаются друг от друга химическим строением – последовательностью связей атомов в молекулах.
Первые три отличаются, соответственно, скелетом, функциональной группой, положением функциональной группы:
|
|
|
|
|
бутан |
диметиловый эфир |
пропиловый спирт |
|
|
|
|
|
изобутан |
этиловый
спирт |
изопропиловый
спирт |
|
а |
б |
в |
Вариантом структурных изомеров, отличающихся скелетом, являются топологические изомеры, о которых уже говорилось ранее. Они могут отличаться размерами, количеством колец в катенанах, колец и гантелей в ротаксанах, элементов узлов и относятся к статическому ряду. Пары г, д являются таутомерами и относятся к динамическому ряду.
|
|
|
||
|
виниловый
спирт |
ацетальдегид |
2-гидроксипиридин |
2-пиридон |
|
г |
д |
||
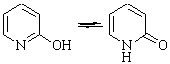
Таутометрия – достаточно распространенное явление в органической химии. Она возможна при переносе подвижного водорода (карбонильные соединения, амины, амиды и др.) внутримолекулярных взаимодействиях (углеводы и др.), валентных превращениях и т. д.
Структурные изомеры отличаются друг от друга химическими и физическими свойствами и могут быть разделены на этой основе обычными методами.
3.2. Пространственная изомерия
Изомеры, отличающиеся друг от друга пространственным расположением атомов в молекуле, называются пространственными.
Раздел химии, изучающий пространственное строение молекул и его влияние на физические и химические свойства, называется стереохимией.
Известны три типа пространственных изомеров, два из которых устойчивы в “нормальных” условиях – геометрические и оптические.
3.2.1. Геометрическая (цис-, транс-) изомерия
Геометрическая изомерия характерна для соединений, имеющих двойную связь. В таких соединениях, как отмечалось ранее, невозможно свободное вращение вокруг двойной связи в „нормальных“ условиях, для этого необходим разрыв p-связи. Изомер, у которого заместители находятся по обе стороны от плоскости двойной связи, называется транс-изомером, если по одну сторону – цис-. Цис- и транс-изомеры отличаются по химическим и физическим свойствам и легко разделяются обычными методами.
|
|
|
|
транс-бутен-2 |
цис-бутен-2 |
Обычно транс-изомеры более стабильны по сравнению с цис-изомерами, хотя известны и обратные случаи.
Геометрическая изомерия наблюдается и в случае двойных связей C=N и N=N. Однако у таких связей изомеризация, то есть взаимопревращение изомеров, происходит значительно легче, чем у двойной связи C=C, поскольку такая изомеризация может осуществляться путем инверсии электронной пары, а не вращения вокруг двойной связи. Примерами таких изомеров могут служить цис- (ранее син-), транс- (ранее анти-) оксимы, азобензолы:
|
|
|
|
цис(син)-оксим |
транс(анти)-оксим |
|
|
|
|
транс-азобензол |
цис-
азобензол |
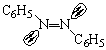
Обычный азобензол является транс-изомером, так как не имеет дипольного момента, но при интенсивном освещении он превращается в более растворимый и более интенсивно окрашенный изомер с дипольным моментом.
3.2.2. Оптическая изомерия
В 1815 г. французский физик Био открыл явление оптической активности – способности жидкостей вращать плоскость поляризации плоскополяризованного света. Обычный свет является совокупностью электромагнитных колебаний с различными длинами волн, колеблющихся во множестве плоскостей, которые перпендикулярны направлению распространения светового луча.
Монохроматический свет, испускаемый, например, натриевой лампой, подобен обычному, но характеризуется одной длиной волны. Если свет пропустить через призму Николя из исландского шпата или кварца, то при выходе он становится поляризованным – электромагнитные колебания происходят в одной плоскости, которая называется плоскостью поляризации. Оптическую активность измеряют приборами, которые называются поляриметрами (рис. 3.1).
Монохроматический свет, пройдя через поляризатор 2 (призма Николя из исландского шпата), становится поляризованным. Если в поляриметрическую трубку 3 поместить раствор с оптически активным веществом, то поляризованный свет, пройдя через него, приобретает плоскость поляризации, смещенную на угол a.
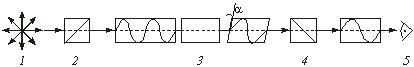
Рис. 3.1. Схема устройства поляриметра:
1 – источник монохроматического
света,
2
– поляризатор,
3 – исследуемый раствор в
поляриметрической трубке,
4 – анализатор,
5 – наблюдатель
Такое смещение наблюдатель будет фиксировать по эффекту полного гашения света поворотом анализатора 4 (призмы Николя расположены таким образом, что плоскости их поляризации повернуты друг к другу на 90 градусов). Измеряют обычно удельное вращение:
![]() , (40)
, (40)
где: [a] – удельное
вращение,
a – угол вращения,
l – длина слоя жидкости в дм,
с – число граммов оптически активного вещества в 100 мл раствора,
l – длина волны монохроматического
света в м,
t – температура в °С.
Возникает вопрос: какие вещества могут обладать оптической активностью?
В 1848 г. Луи Пастер, отделив пинцетом друг от друга кристаллы двух зеркальных форм оптически неактивной натрий-аммониевой соли виноградной кислоты, получил из неактивной смеси оптически активные вещества. Оба изомера смещали плоскость поляризации поляризованного света на один и тот же угол, но в разные стороны: один – по часовой стрелке (правовращающий, условно (+)), другой – против часовой стрелки (левовращающий, условно (–)). Так впервые были получены право- и левовращающие винные кислоты из неактивной виноградной кислоты. Эти изомеры были названы оптическими антиподами, или оптическими изомерами.
3.2.2.1. Термины, используемые
при рассмотрении
оптической изомерии
Оптические изомеры – пространственные изомеры, имеющие одинаковый количественный и качественный состав, одинаковое химическое строение, идентичные по всем физическим и химическим свойствам, но отличающиеся способностью вращения плоскополяризованного света, способностью образовывать при кристаллизации кристаллы, являющиеся зеркальными отражениями друг друга.
Оптическая изомерия является важным свойством органических соединений, которое оказывает существенное влияние на их свойства. Например, такие важные природные соединения, как углеводы, белки, оптически активны.
Пастер сформулировал причину возникновения оптической активности следующим образом: “...расположены ли атомы винной кислоты подобно резьбе правого винта, находятся ли они по углам неправильного тетраэдра или образуют какую-либо другую асимметрическую группировку, мы не можем ответить на этот вопрос. Но вне всякого сомнения, что атомы в молекуле винной кислоты обладают несимметричным расположением по типу предмета и его зеркального изображения, не способных к взаимному совмещению...” [22]. Гениальная догадка Пастера, высказанная еще до теории химического строения А. М. Бутлерова, была подтверждена и развита стереохимической теорией Вант-Гоффа и Ле Беля в 1874 г., работами современных исследователей.
Оптическая изомерия в химии является частным случаем проявления общего свойства природы – симметрии. Правая и левая руки, перчатки для правой и левой руки, правая и левая половины листа, тела и др. характерны тем, что пары – объект и его зеркальное отражение – не совместимы друг с другом.
Рассмотрим некоторые необходимые термины.
Конфигурация – расположение атомов или групп атомов заместителей в пространстве без учета ориентации заместителей, связанной с поворотом вокруг ординарной связи.
Плоскость симметрии – плоскость, которая рассекает объекты таким образом, что обе части относятся друг к другу как предмет и его зеркальное изображение (они могут быть совместимыми и несовместимыми друг с другом).
В зависимости от строения молекулы ей может соответствовать одна или несколько плоскостей симметрии.
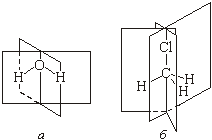
Вода (а) имеет две плоскости симметрии, хлорметил (б) – три. Все плоские молекулы имеют по крайней мере одну плоскость симметрии, линейные – бесконечное множество.
Ось симметрии – линия, вращение молекулы вокруг которой на угол 2p/n = 360°/n приводит к структуре, идентичной исходной, называют осью симметрии n-го порядка Сn. Естественно, условию n = 1, то есть когда угол вращения равен 360°, удовлетворяют любые молекулы:
|
|
|
|
|
|
а |
б |
в |
г |
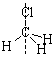
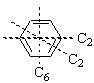
Молекула воды (а) имеет ось симметрии второго порядка С2, хлорметана (б) – третьего порядка С3, бензола (в) – шестого порядка С6 и второго порядка С2, ацетилена (г) – бесконечного порядка С¥. Ось симметрии самого высокого порядка называют главной, или осью сравнения.
Центр симметрии. Точка в центре молекулы, относительно которой каждому атому в молекуле соответствует эквивалентный атом по линии, проходящей через эти атомы и центр молекулы, называется центром симметрии. Бензол (а) и этилен (б) – примеры молекул с центрами симметрии:
|
|
|
|
|
|
|
а |
б |
|
Хиральность – способность соединений существовать в виде пары несовместимых между собой зеркальных изображений. Если молекула не имеет ни центра, ни плоскости симметрии, то она хиральна, если имеет, то она ахиральна, не имеет энантиомеров.
Хиральный центр – обычно атом с
четырьмя различными заместителями (асимметрический).
Энантиомеры – любые пары объекта и его изображения, несовместимые друг с другом. Соединение называется хиральным, если оно существует в виде пары энантиомеров, таким образом, хиральность – необходимое и достаточное условие для существования энантиомеров.
Рацемат, рацемическая форма – эквимолекулярная смесь энантиомеров, не обладающая оптической активностью вследствие внутренней взаимной компенсации, обозначается (±).
Диастереомеры – стерео(пространственные)изомеры, не являющиеся энантиомерами, то есть зеркальными изображениями друг друга, отличаются друг от друга физическими и химическими свойствами.
3.2.2.2. Структуры, способные существовать в виде энантиомеров
Первый случай. Молекула имеет sp3-гибридизованный атом с четырьмя разными атомами или группами атомов. Такой центральный атом называют хиральным или асимметрическим:
|
|
|
|
|
|
|
а |
|
б |
||
|
|
|
|
|
|
|
в |
|
г |
||
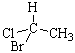
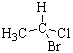
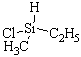
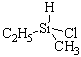
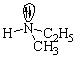
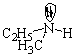
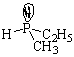
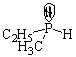
В парах а, б, в, г хиральные атомы – соответственно, углерод, кремний, азот, фосфор, причем два последних в качестве четвертого заместителя имеют неподеленные пары электронов.
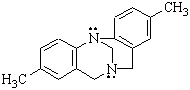
Теоретически возможные пары в и г на самом деле не существуют, так как пирамидальная структура азота и фосфора очень легко претерпевает инверсию – переход одной конфигурации в другую. Однако, если затруднить инверсию, то энантиомеры возможны, например, в “основании Трегера”:
Таким образом, оптическая активность возможна лишь тогда, когда соединение хирально и конфигурационно устойчиво.
В отличие от соединений трехвалентного фосфора соединения пятивалентного фосфора, содержащие фосфор в sp3d-гибридизованном состоянии (тригональная бипирамида), дают устойчивые энантиомеры, например, пентазамещенные производные фосфора:
Второй случай. Еще в 1875 году Вант-Гофф предсказал возможность существования энантиомерных форм несимметрично замещенных алленов типа 1, 2, 3:
|
|
|
|
|
тип 1 |
тип 2 |
тип 3 |
Такая возможность следовала из стереохимической модели строения алленов, согласно которой заместители a, b y атомов C1 и С3 располагаются во взаимно перпендикулярных (ортогональных) плоскостях.
|
|
или |
|
Если по Вант-Гоффу модели СН4 и СН2=СН2 представить, соответственно, в виде тетраэдра и двух тетраэдров, связанных ребром, то аллен типа 1 примет вид косоугольного тетраэдра, способного существовать в виде двух энантиомеров, являющихся зеркальными отражениями друг друга:
|
|
||
|
CH4 |
C2H4 |
|
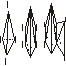
Впервые оптически активные аллены получили в 1936 г. Мильс и Мейтленд.
Дегидратация 1,3-дифенил-1,3-ди(a-нафтил)пропен-2-ола-1 в присутствии (+)- или (–)-камфор-10-сульфокислоты
(КСК) дает (+)-
и (–)-1,3-дифенил-1,3-ди(a-нафтил)аллены
соответственно.
|
|
||
|
1,3-дифенил-1,3-ди-(a-нафтил)-пропен-2-ол-1 |
|
(+)-1,3-дифенил-1,3-ди- |
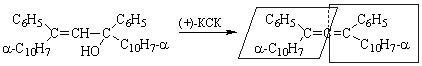
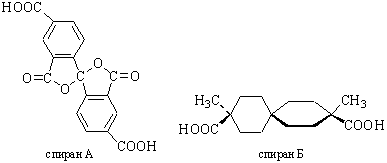
Третий случай. Вант-Гофф предсказал существование энантиомерных форм и для спиранов – бициклических соединений с атомом углерода, принадлежащим обоим циклам одновременно. Если принять упрощенно циклогексановые кольца плоскими, то возможна ситуация, аналогичная алленам типа 1, 2, 3, если присутствуют заместители, создающие ассиметрию, например:
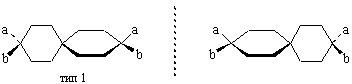
Первый оптически активный спиран А описан в1920 г. Мильсом и Нодером, а в 1928 г. Безекен получил спиран Б.
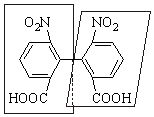
Четвертый случай. Оптическая изомерия возможна и у бифенилов с четырьмя объемными заместителями в орто-положениях. В таких случаях свободное вращение вокруг ординарной связи затруднено из-за пространственых препятствий при вращении, и удается выделить два энантиомера, если в о,о-положениях расположены разные заместители (по два разных в кольце или все четыре разные). Примером таких энантиомеров могут служить 6,6'-динитродифеновые кислоты.
|
|
|
|
(+)-6,6'-динитродифеновая кислота |
(–)-6,6'-динитродифеновая кислота |
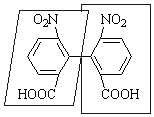
Оптическая изомерия, связанная с затрудненным свободным вращением (атропоизомерия), возможна и для бинафталинов, бифенантренов и т. д. Атропоизомерия может рассматриваться как предельный случай конформационной изомерии (см. далее), но резкого перехода между ними нет. Если при комнатной температуре энергетический барьер взаимоперехода атропомеров составляет 67–84 кДж/моль и позволяет выделить их, то при повышенных температурах взаимопереход ускоряется и их выделение становится невозможным, как и конформационных изомеров (см. далее).
Более подробно вопросы стереохимии соединений углерода см. в [22–24].
3.2.2.3. Способы изображения трехмерных объектов
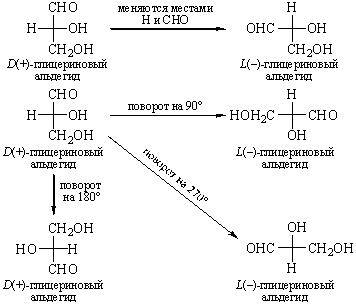
Проекции Фишера. Хиральный центр рисуют с четырьмя связями, расположенными под прямым углом друг к другу. Вертикальные линии изображают заместителей, находящихся за плоскостью листа бумаги, горизонтальные – над плоскостью, центральный атом находится в плоскости листа. Атом углерода в качестве центрального обычно не изображается, другие атомы изображаются. Хиральный (асимметрический) атом часто помечают звездочкой *.
Если в проекции Фишера поменять местами соседние группы, то получают зеркальное изображение исходного соединения. Поворот всей молекулы на 90° и 270° дает одинаковый результат. Поворот всей молекулы на 180° и 360° приводит к первоначальной структуре:
Перспективные формулы – модификация проекций Фишера, в которой горизонтальные линии рисуются жирными клиньями, вертикальные – штриховыми клиньями.
Тетраэдрическое изображение – перспективные формулы в тетраэдрическом виде.
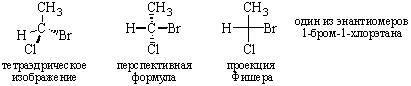
3.2.2.4. Относительная и абсолютная конфигурация
Поляриметр позволяет определить только знак вращения, но не указывает, какой конкретно энантиомер имеет такое вращение. Для этого необходимо определить абсолютную конфигурацию, то есть истинное расположение заместителей в пространстве относительно хирального центра. В настоящее время используют D – L номенклатуру (в основном для углеводов и аминокислот) и R – S номенклатуру Кана – Ингольда – Прелога, которая носит универсальный характер.
В качестве стандарта при использовании D – L номенклатуры Э. Фишер предложил D(+)-глюкозу, а М. Розанов в 1906 г. – D(+)-глицериновый альдегид, которому условно приписали расположение заместителей:
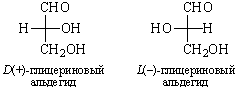
В 1951 г. Бийо установил, что конфигурация D-глицеринового альдегида именно такая. Абсолютную конфигурацию других соединений определяют относительно этого стандарта (см. главу XXIII).
Абсолютные конфигурации оптических изомеров относятся к двум рядам D и L как производные эталонов, то есть D- и L-глицериновых альдегидов. Знак вращения и принадлежность к D- и L-ряду (или R- и S-ряду) не совпадают, знак определяется в каждом случае экспериментально. Если два или несколько соединений имеют три заместителя, одинаково ориентированных вокруг четвертого, то говорят, что такие соединения имеют одинаковую относительную конфигурацию (например, эпимеры в углеводах, см. главу XXIII).
Определение абсолютной конфигурации соединения его синтезом
из оптически активного соединения с заведомо известной абсолютной конфигурацией
– обычно длительный и трудоемкий процесс. Наиболее приемлемым из известных
методов выражения абсолютных конфигураций является метод Кана – Ингольда –
Прелога. В соответствии
с номенклатурой Кана – Ингольда – Прелога, называемой R – S-системой, четыре
заместителя хирального центра располагаются в порядке старшинства,
определяемого правилами (см. ниже), и абсолютная конфигурация хирального центра
определяется согласно рис. 3.2.
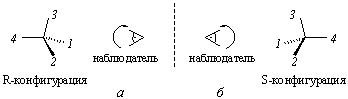
Рис.
3.2. Определение абсолютной конфигурации хирального центра
1 > 2 > 3 > 4, где заместитель 4 – самый младший
Если наблюдатель располагается по линии, связывающей младший заместитель (4) с хиральным центром с противоположной этому заместителю стороны и при движении по часовой стрелке видит последовательность 1®2®3 (рис. 14, а), то конфигурация хирального центра обозначается R (от латинского rectus – правый), против часовой стрелки (рис. 14, б) – символом S (от латинского sinister – левый). Рацемическая форма обозначается символом R,S. Например, энантиомер (R)-1-бром-1-хлорэтан, рацемическая форма (R,S)-1-бром-1-хлорэтан.
Правила старшинства в системе Кана – Ингольда – Прелога:
1. Атом или атом заместителя, непосредственно связанный с хиральным центром, с бόльшим атомным номером является старшим относительно атома с меньшим атомным номером.
2. Большее массовое число имеет преимущество перед меньшим, в том числе и для изотопов.
3. Неподеленные пары электронов имеют самый низкий порядок, в том числе и по сравнению с водородом.
4. Если с хиральным центром связаны два идентичных атома, то старшим считается тот, который связан с атомом с большим атомным номером; процедура может повторяться.
5. Если атом двоесвязан, то по старшинству он приравнивается
к атому, связанному с двумя эквивалентными группами. Так, группа ![]() рассматривается как
рассматривается как ![]() , где (О) и (С) – атомы, идентичные находящимся на конце
двойной связи.
, где (О) и (С) – атомы, идентичные находящимся на конце
двойной связи.
Примеры определения абсолютной конфигурации:
|
|
|
|
|
|
|
|
|
а |
б |
|
|
|
|
||
|
|
|
||
|
в |
г |
||
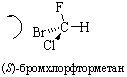
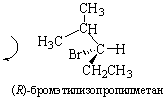
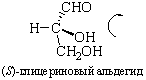
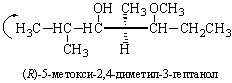
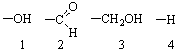
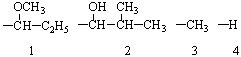
В случае б атом углерода в изопропильной группе связан с двумя углеродами по сравнению с одним в этильной группе.
В случае в углерод альдегидной группы двоесвязан с кислородом.
В случае г кислород метоксиалкильного заместителя связан с двумя углеродами.
Таким образом, можно расположить атомы и заместители, содержащие атом, который непосредственно связан с хиральным центром, в порядке уменьшения старшинства следующим образом:
![]()
![]()
![]()
![]()
![]()
![]()
Подробно о системе Кана – Ингольда – Прелога см. [25].
Практика перехода от D – L номенклатуры к R – S рассмотрена в следующем разделе.
3.2.2.5. Соединения с несколькими хиральными центрами
В соединении Сabc–Cabc имеются два хиральных центра (асимметрических атома) (С), поэтому возможны следующие четыре структуры, где R или S – абсолютные конфигурации хиральных центров С:
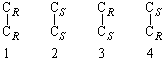
Соединения 1 и 2 являются энантиомерами, то есть несовместимыми зеркальными изображениями.
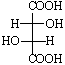
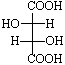
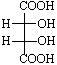
Структуры 3 и 4 идентичны, а пары соединений 1 и 3, 2 и 3 являются диастереомерами по отношению друг к другу. Примерами таких соединений служат винные кислоты, свойства которых хорошо иллюстрируют разницу в свойствах энантиомеров и диастереомеров (табл. 3-2).
В R – S номенклатуре винные кислоты описываются следующим образом:
|
|
|
||||
|
D(+)-винная |
(2R,3R)(+)-винная
кислота |
L(–)-винная кислота |
(2S,3S)(–)-винная кислота |
||
|
|
|||||
|
|
мезо-винная
кислота |
(2R,3S)-винная
кислота |
|
||
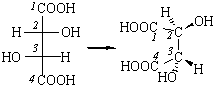
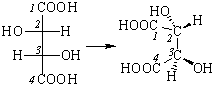
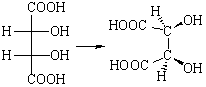
В тетраэдрических изображениях винных кислот атомы углерода располагаются в одной плоскости.
Отличительным признаком мезо-винной кислоты (мезо-форм в общем виде) является наличие в одной молекуле двух противоположных энантиомерных фрагментов, взаимно компенсирующих оптическое вращение (2R и 3S хиральные центры в мезо-винной кислоте). Так как оптические изомеры с одним хиральным центром конфигурационно устойчивы, то есть D- (или R-) изомеры не превращаются в L- (или S-) изомеры, аналогично мезо-форма конфигурационно устойчива и не должна превращаться ни в D- (R-), ни в L- (S-) форму (для одного из хираль ных центров второй хиральный центр входит в состав одного из его заместителей!).
Таблица
3-2
Винные кислоты
|
Формула |
|
|
|
|
|
Название |
D(+)-винная |
L(–)-винная |
мезовинная |
виноградная |
|
Т. пл., °С |
170 |
170 |
140 |
206 |
|
|
+12° |
–12° |
±0° |
±0° |
|
Растворимость, |
139 |
139 |
125 |
20,6 |
|
К1×103 |
1,17 |
1,17 |
0,77 |
1,10 |
|
К2×105 |
5,9 |
5,9 |
1,6 |
5,8 |
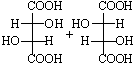
Теперь
расмотрим более сложный случай – соединения типа
Cabc–Cabd, для которых
тоже возможны четыре структуры, где B = Cabс и D = Cabd:
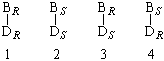
В данном случае пары 1 и 2, 3 и 4 являются энантиомерами, то есть несовместимыми зеркальными изображениями, а пары 1 и 3, 1 и 4, 2 и 3, 2 и 4 – диастереомерами, то есть не являются зеркальными изображениями.
В энантиомерных парах 1 и 2, 3 и 4 возможны два случая: все идентичные группы (a, b) заслоняют друг друга – это эритро-формы:
|
|
эритро-энантиомеры |
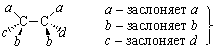
или заслонена максимум одна пара идентичных заместителей (а и а) – это трео-формы:
|
|
трео-энантиомеры |
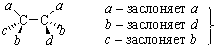
Примеры эритро- и трео-форм:
|
|
|
|
|
|
трео-формы |
|
||
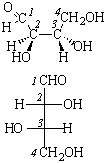
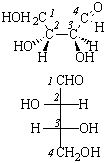
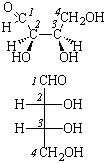
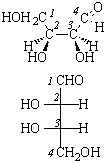
Соединения типа Сabc–Cdef существуют в виде двух пар зеркальных изображений, то есть каждый хиральный центр как бы независим от другого, другой хиральный центр входит в состав одного из заместителей первого.
|
|
|
|
|
|
1 |
2 |
3 |
4 |
Соединения 1 и 2, 3 и 4 являются энантиомерами, а пары 1 и 3, 2 и 3, 1 и 4, 2 и 4 – парами диастереомеров.
С увеличением числа хиральных центров число стереоизомеров растет. Для молекул с n числом неодинаковых хиральных центров число возможных стереоизомеров составляет
N = 2n, (41)
где: N – общее число возможных стереоизомеров,
n – число хиральных
центров.
Структура конформеров часто оказывает решающее влияние на физические и химические свойства соединений, поэтому так интенсивны исследования в этой области в настоящее время.
Пример этого свойства конформационных изомеров циклогексана рассматривается в главе XIII.
3.2.3. Конформационная изомерия
К динамическому ряду пространственных изомеров относятся конформационные изомеры, или конформации. Появление конформаций связано с тем, что хотя вокруг ординарной связи должно быть свободное вращение, однако вследствие взаимного отталкивания атомов и групп атомов существует некоторый энергетический барьер, который необходимо преодолеть для осуществления такого свободного вращения. Энергетический профиль вращения вокруг связи С–С этана и проекции Ньюмена конформаций последнего представлены на рис. 3.3.
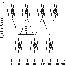
Рис. 3.3. Энергетический профиль вращения вокруг связи С–С этана
Проекции Ньюмена получают, рассматривая соединение вдоль ординарной связи, например, С–С связи этана:
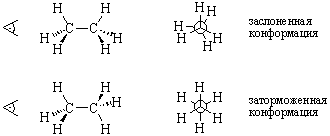
Энергетический барьер между заслоненной и заторможенной
конформациями (барьер вращения) этана равен 12,5 кДж/моль (для предотвращения
вращения требуется энергетический барьер порядка 84–130 кДж/моль), то есть
теплового движения молекул при комнатной температуре с избытком хватает для
преодоления этого барьера. По этой причине пространственные изомеры этана
находятся в динамическом равновесии и их невозможно выделить в индивидуальном
виде при “нормальных” условиях. Такие пространственные изомеры называют конформационными, или конформациями. Ниже приведены барьеры
вращения (кДж/моль) вокруг некоторых ординарных связей приводятся ниже [26, с.
71]:
|
CH3–CH2F |
13,81 |
CH3–OH |
4,48 |
|
CH3–CH2Cl |
14,89 |
CH3–SH |
5,27 |
|
CH3–CH2Br |
14,94 |
CH3–NH2 |
8,12 |
|
|
8,28 |
|
2,01 |
|
|
83,68 |
|
29,29 |
Барьер вращения существенно выше при наличии сопряжения, что связано с частично двоесвязанным характером связей C–N и C–C у ацетамида и акролеина соответственно.
Конформации, обладающие минимумом энергии и наиболее стабильные по этой причине, называются конформерами, например, заторможенные конформации этана.
Свободному вращению вокруг ординарной связи препятствуют при сближении групп наряду со взаимным ван-дер-ваальсовым отталкиванием изменения:
– торсионного (питцеровского) напряжения (квантово-химическое взаимодействие соседних s-связей, например, в конформации “ванна” циклогексана);
– углового или байеровского напряжения (в циклоалканах);
– дипольных взаимодействий или водородных связей;
– энергии сольватации (в растворителе при смене растворителя);
– напряжения связей вследствие их растяжения или сжатия (при изменении температуры);
– несвязных взаимодействий.
Энергетические барьеры свободного вращения вокруг ординарной связи могут варьировать поэтому в широких пределах.
Задача определения
равновесных взаимопревращений конформаций и структуры стабильных
конформеров зависит, таким образом, от многих факторов и решается различными, в
том числе физико-химическими методами; эта область химии называется конформационным анализом [24].